
Pedro Camargo, Ph.D.
Tuesday, October 16, 2018
Biogeme on Windows using Docker
An important part of my day job is to develop new transportation models, and that includes a lot of discrete choice model estimation, and in the last 18 months that has included anywhere from simple departure time choice models, to more sophisticated mode choice models, where we experimented with Nested, Cross-Nested and Mixed-Logit. We have also estimated standard destination choice models based on standard MNL, but with close to 1,000 alternatives and with utility functions that are non-linear in parameters, which makes the choice sets incredibly large and the estimation very time consuming.
When you combine this less-than-trivial model estimation with a highly segmented model, you are left with the need to estimate and analyze many hundreds of models, so manually preparing choice sets, estimation scripts and outputs for analysis is out of question. The tooling for doing such is what we (as in literally our team) call machinery , and it is done very much with a software development mindset, including unit tests, version controlling cross-checking on pull/merge-requests. It takes time, but the gain in productivity is just astounding.
There are many great software out there that are great at discrete choice model estimation, both proprietary and open source, but working in this “intensive model development”environment scenario restricts the software we can actually use, mostly because of their APIs and/or command line capabilities. We also restricted ourselves to software that has already been thoroughly checked and its results compared to Monte-Carlo studies, which rules out a number of fairly interesting open-source packages, especially in the Python and R ecosystems.
For these (and some other) reasons, we have chosen BIOGEME a while, and we are pretty happy with. The possibility of using Python as its scripting language allows us to integrate BIOGEME in our model development workflow and leverage our Python skills, which is great. It can also be run on the command line, which allows us to integrate a lot of Bash scripting as well.
There is a problem, though: BIOGEME for Windows is really bad. I mean… It is ATROCIOUS.
Because of that, we adopted a workflow of utilizing an Ubuntu Virtual Machine for model estimation, and I wrote a post about it, where I made the virtual box available (it is still there). Since then I have migrated to a laptop with a dual boot Windows/Ubuntu, and end up working on Linux every time I am estimating models, but my team still works mostly on Windows, as most of our software is Windows only. The Linux VM has become a burden, however, and while the whole team would prefer to just move to Linux, we have to work on Windows for many important reasons.
But what if we could run Biogeme on Windows properly?
Enter Docker.
First of all, while I didn’t know if it would work, I was definitely not the first one to go for it, but I couldn’t find many recent work done on it, tutorials or any proper documentation, hence this blog post. The desired setup had simple objective: Run BIOGEME on Windows with the least amount of Docker setup and knowledge possible.
I put together a small repo on GitHub with an example of how to use Docker on Windows to run all the Swiss Metro examples provided by Bierlaire, which started as a fork of an outdated Python Biogeme Docker image (reference on GitHub). In any case, the gist is the following:
Docker requires you to register in order to download it, and to login in your local computer, which is a tad annoying, but nothing major.
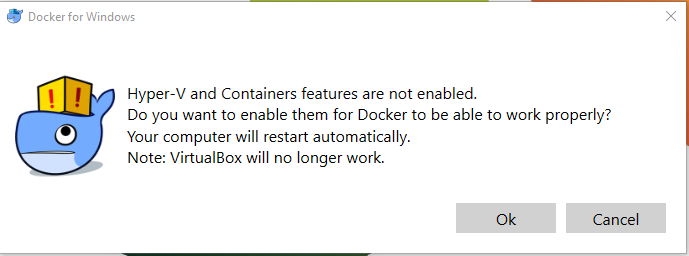
There are plenty resources on this topic online (I linked only one of them here), but you need to be aware that you will get some warnings like a request to activate Hyper-V, which will automatically reboot your PC
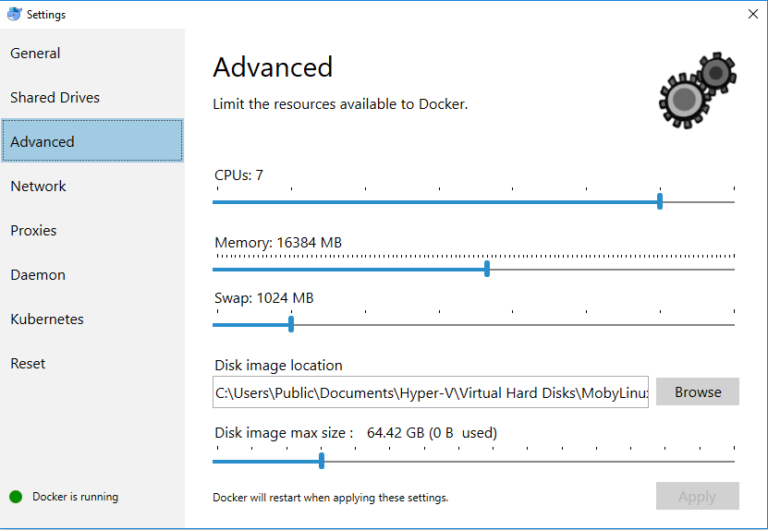
- Configuring Docker (in order to have enough CPU and memory to run more intensive BIOGEME jobs)
3. Create a Biogeme Docker image
This is just a single command once you clone/download the repository and navigate to the repo folder with your command line software: docker build -t biogeme .
4. Estimate away!
Running BIOGEME consists of running a single command from your command prompt: docker run -v D:/BIOGEME://tmp/mr -w //tmp/mr biogeme pythonbiogeme estimate dataset.dat
Where the blue part is just telling Docker to run by sharing a host folder with the docker machine, to start in that shared folder (or the subfolder that contains the scripts), running the docker image that has that name, and calling the standard Python BIOGEME command.
Finally, if you can work 100% of the time on Linux, as I have been able to do, go a head and do it. If not, Docker does provide a seamless way forward on Windows.
The full video tutorial is here